

What is HAE?

HAE-A rare genetic disease
Hereditary angioedema (HAE) is a rare and potentially life-threatening genetic disease affecting ~1:50,000 people. Individuals with HAE experience recurring and unpredictable cutaneous and submucosal oedema of various body sites.1, 2
The cause of HAE
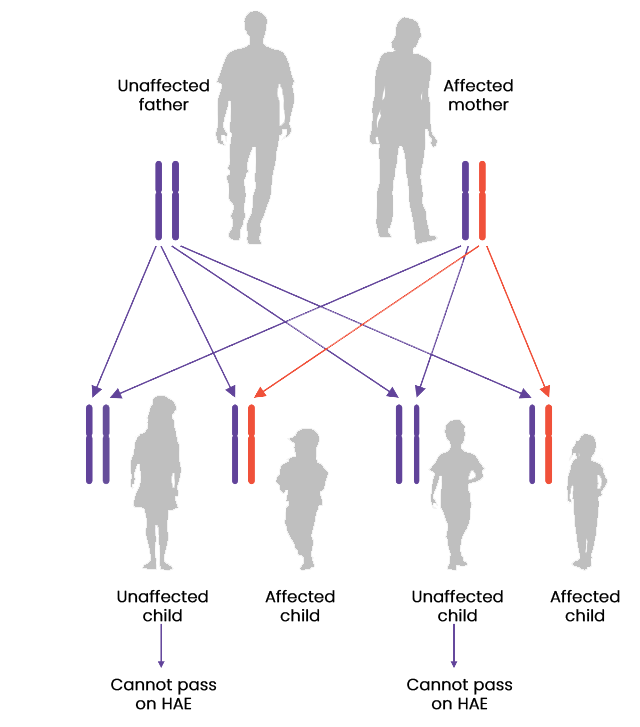
Most cases of HAE are caused by a mutation in the SERPING1 gene which encodes the protein C1 esterase inhibitor (C1-INH), resulting in either a deficiency (type I) or dysfunction (type II) of C1-INH. HAE is an autosomal dominant disease; therefore, it is recommend that family members and offspring of diagnosed patients are also tested for HAE. However, 25% of cases occur due to de novo mutations.1, 3
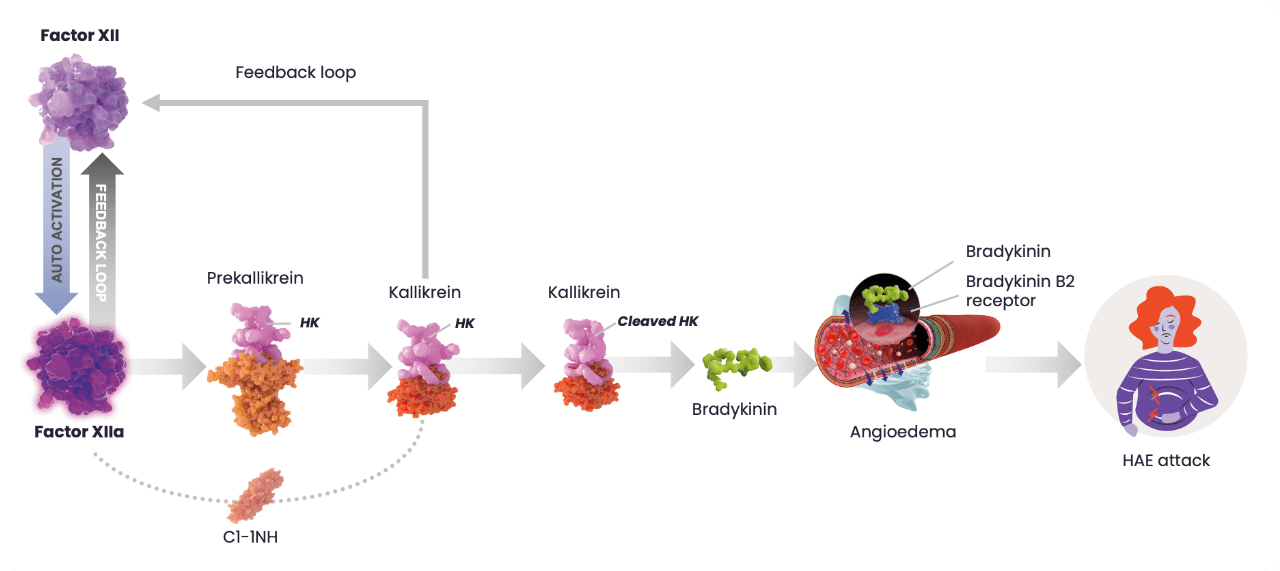
The HAE cascade
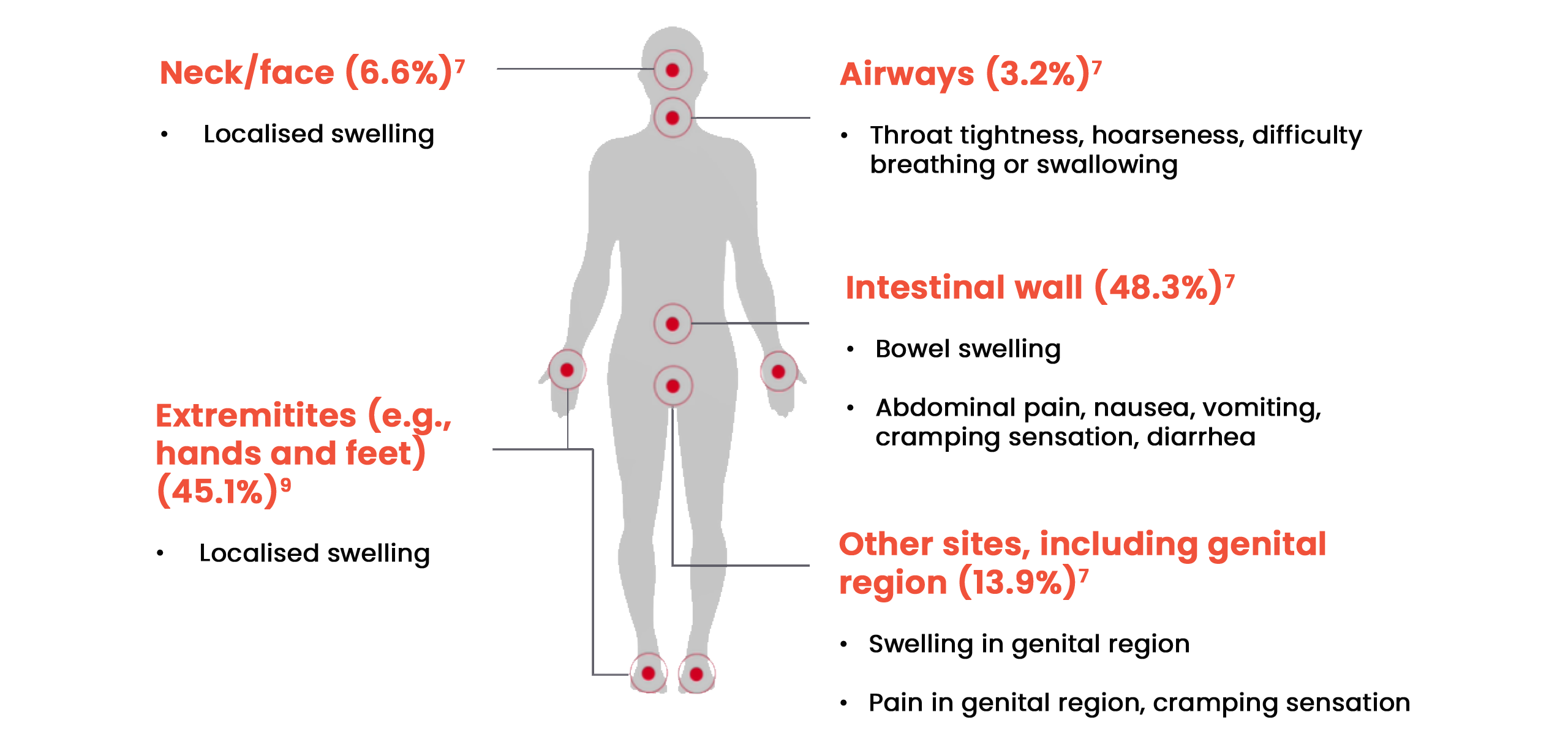
Attack locations
Patients can experience symptoms in a range of locations on the body:7,8,9
Laryngeal oedema
Laryngeal oedema requires emergency attention, and can be life-threatening, particularly in patients who have not yet been diagnosed with HAE.14 Patients should be made aware of the signs of a laryngeal attack, which include:9
- Swallowing difficulties
- Tightness or a feeling of a “lump in the throat”
- Shortness of breath
- Vocal changes such as loss of voice, hoarseness or a rough voice

Prodromal symptoms
Prodromal symptoms act as signs of a potential attack and can vary between each person. Common indicating symptoms include:15

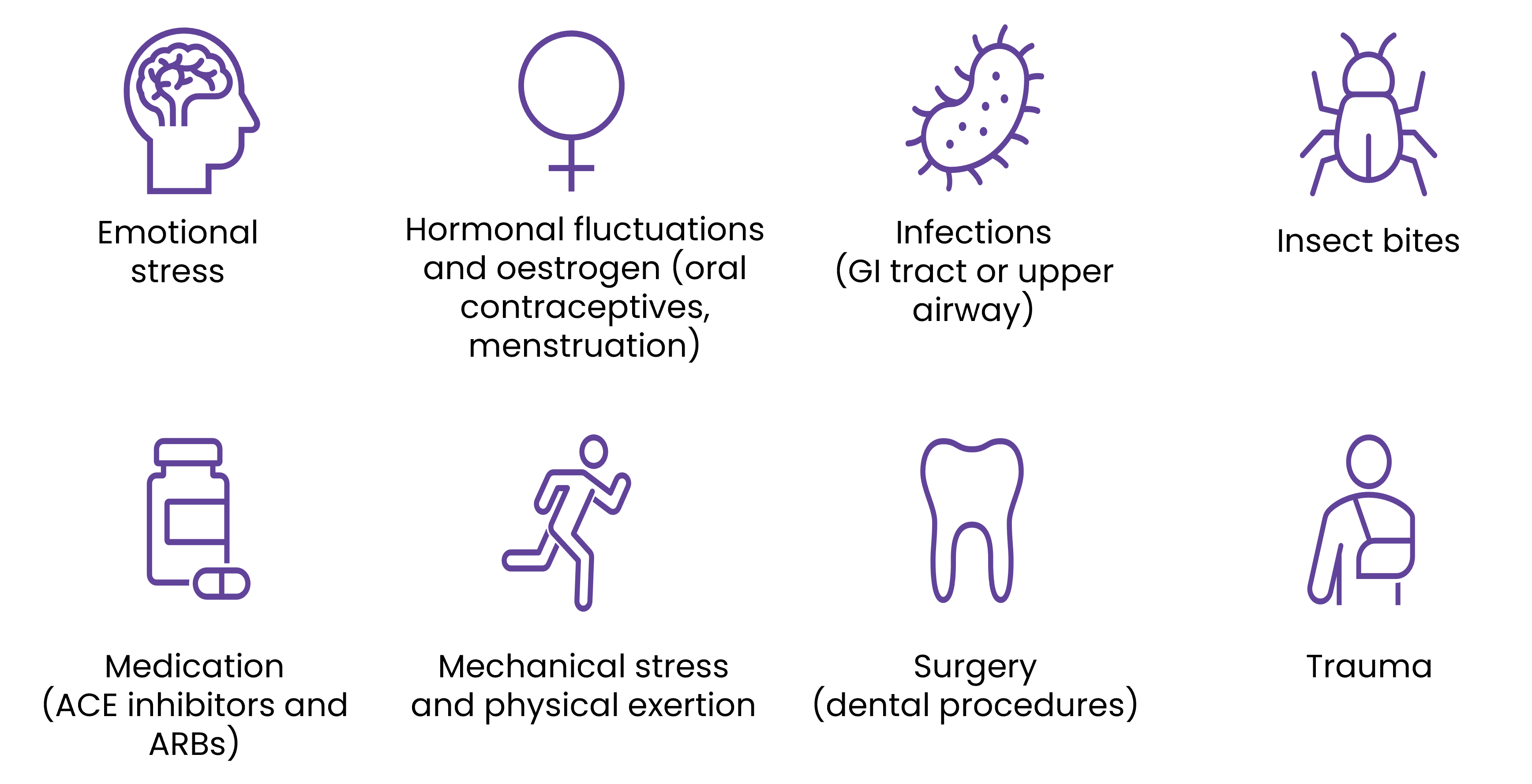
Potential triggers
While most HAE attacks happen spontaneously and unexpectedly, there are various triggers that can cause an attack; however these may be specific for each patient with HAE.17 Some of the common triggers that may affect patients include:1,17-19
Diagnosis of HAE
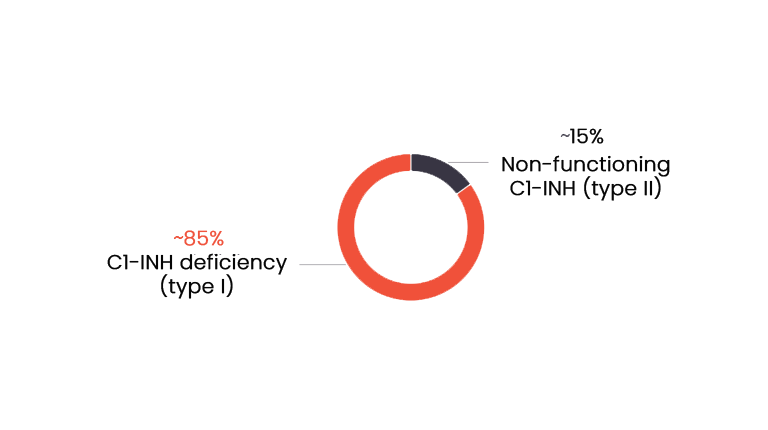
Types of HAE
In most cases, HAE is caused by deficiency (type I) or dysfunction (type II) of C1-INH, that results in uncontrolled activation of the contact system and release of bradykinin.3
However, HAE can also be diagnosed in patients with normal C1-INH due to mutations in other genes. People diagnosed with HAE with normal C1-INH (HAE-nC1-INH) experience largely similar clinical symptoms as those with type I or II HAE.1
HAE-nC1-INH is classified by the gene in which the mutation occurs.
Currently there are 7 known HAE-nC1-INH types classified by the gene in which the mutation occurs;20,21
- HAE-FXII
- HAE-PLG (plasminogen)
- HAE-KNG (kininogen 1)
- HAE- HSST (heparan sulfate glucosamine 3-O-sulfotransferase 6)
- HAE- ANGPT (angiopoietin-1)
- HAE-MYOF (myoferlin)
- HAE-CPN (carboxypeptidase N)
- HAE-Unknown
The most common type of HAE-nC1-INH is HAE-FXII, which has been reported in 20-25% of patients with HAE-nC1-INH.22 The mediator of angioedema varies between types of HAE-nC1-INH, and in some cases is still unknown.1,23
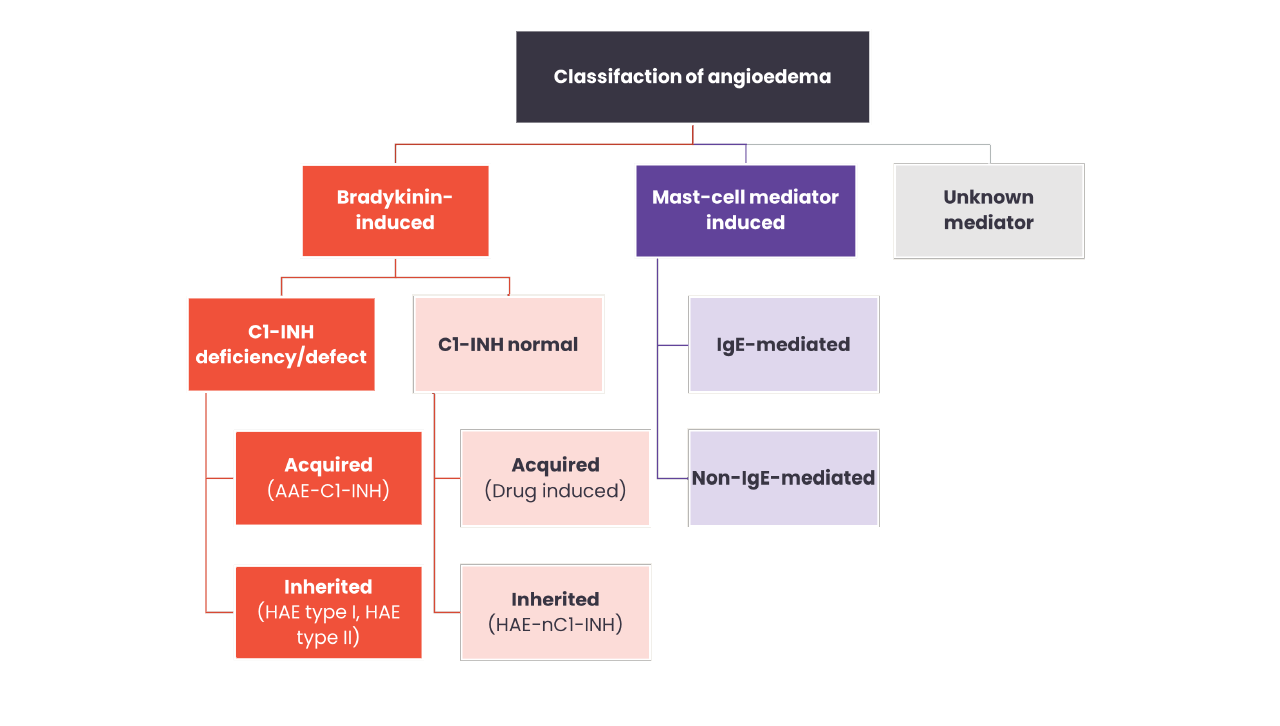
Click here for a detailed table summarising the key differences between angioedema types
Early diagnosis is vital!
Early and correct diagnosis of HAE is key, especially due to the potential for laryngeal swellings, which pose a risk of asphyxiation.14
When should HAE be suspected?1
- Positive family history (75% of patients)
- Onset of symptoms in childhood/adolescence
- Recurrent and painful abdominal symptoms
- Occurrence of upper airway oedema
- Failure to respond to antihistamines, glucocorticoids, omalizumab or epinephrine
- Presence of prodromal signs or symptoms before swellings
- Absence of wheals
Laboratory tests for HAE
After clinical suspicion has been raised, a simple blood test can be used to diagnose HAE by assessing:1

If tests confirm a diagnosis of HAE, it is recommended that the whole family also be tested, due to the genetic nature of the disease. The earlier HAE can be diagnosed, the quicker a suitable management plan can be implemented for the patient.1

Goals of HAE treatment and available treatment options
Find out the goals of HAE treatment and treatment recommendations

Considerations for special patient populations
Learn how to manage HAE in special populations
References
- Maurer M, Magerl M, Betschel S, Aberer W, Ansotegui IJ, Aygören-Pürsün E, et al. The international WAO/EAACI guideline for the management of hereditary angioedema-The 2021 revision and update. Allergy. 2022;77(7):1961–90.
- Bork K, Anderson JT, Caballero T, Craig T, Johnston DT, Li HH, et al. Assessment and management of disease burden and quality of life in patients with hereditary angioedema: a consensus report. Allergy Asthma Clin Immunol. 2021;17(1):40.
- Guryanova I, Suffritti C, Parolin D, Zanichelli A, Ishchanka N, Polyakova E, et al. Hereditary angioedema due to C1 inhibitor deficiency in Belarus: epidemiology, access to diagnosis and seven novel mutations in SERPING1 gene. Clin Mol Allergy. 2021;19(1):3.
- Caccia S, Suffritti C, Cicardi M. Pathophysiology of Hereditary Angioedema. Pediatr Allergy Immunol Pulmonol. 2014;27(4):159-63.
- Kaplan AP, Joseph K. The bradykinin-forming cascade and its role in hereditary angioedema. Ann Allergy Asthma Immunol. 2010;104(3):193-204.
- Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med. 2008;359(10):1027-36.
- Zanichelli A, Farkas H, Bouillet L, Bara N, Germenis AE, Psarros F, et al. The Global Registry for Hereditary Angioedema due to C1-Inhibitor Deficiency. Clin Rev Allergy Immunol. 2021;61(1):77-83.
- Azmy V, Brooks JP, Hsu FI. Clinical presentation of hereditary angioedema. Allergy Asthma Proc. 2020;41(Suppl 1):S18-s21.
- Bork K, Meng G, Staubach P, Hardt J. Hereditary Angioedema: New Findings Concerning Symptoms, Affected Organs, and Course. Am J Med. 2006;119(3):267-74.
- Agostoni A, Aygören-Pürsün E, Binkley KE, Blanch A, Bork K, Bouillet L, et al. Hereditary and acquired angioedema: problems and progress: proceedings of the third C1 esterase inhibitor deficiency workshop and beyond. J Allergy Clin Immunol. 2004;114(3 Suppl):S51-131.
- Mendivil J, Murphy R, de la Cruz M, Janssen E, Boysen HB, Jain G, et al. Clinical characteristics and burden of illness in patients with hereditary angioedema: findings from a multinational patient survey. Orphanet J Rare Dis. 2021;16(1):94.
- Zanichelli A, Longhurst HJ, Maurer M, Bouillet L, Aberer W, Fabien V, et al. Misdiagnosis trends in patients with hereditary angioedema from the real-world clinical setting. Ann Allergy Asthma Immunol. 2016;117(4):394-8.
- Koruth JS, Eckardt AJ, Levey JM. Hereditary angioedema involving the colon: endoscopic appearance and review of GI manifestations. Gastrointestinal Endoscopy. 2005;61(7):907-11.
- Bork K, Hardt J, Witzke G. Fatal laryngeal attacks and mortality in hereditary angioedema due to C1-INH deficiency. J Allergy Clin Immunol. 2012;130(3):692-7.
- Magerl M, Doumoulakis G, Kalkounou I, Weller K, Church MK, Kreuz W, et al. Characterization of prodromal symptoms in a large population of patients with hereditary angio-oedema. Clin Exp
- Dermatol. 2014;39(3):298-303.
- Farkas H, Harmat G, Fáy A, Fekete B, Karádi I, Visy B, et al. Erythema marginatum preceding an acute oedematous attack of hereditary angioneurotic oedema. Acta Derm Venereol. 2001;81(5):376-7.
- Zotter Z, Csuka D, Szabó E, Czaller I, Nébenführer Z, Temesszentandrási G, et al. The influence of trigger factors on hereditary angioedema due to C1-inhibitor deficiency. Orphanet J Rare Dis. 2014;9:44.
- Bowen T, Cicardi M, Bork K, Zuraw B, Frank M, Ritchie B, et al. Hereditary angiodema: a current state-of-the-art review, VII: Canadian Hungarian 2007 International Consensus Algorithm for the Diagnosis, Therapy, and Management of Hereditary Angioedema. Ann Allergy Asthma Immunol. 2008;100(1 Suppl 2):S30-40.
- Maurer M, Aygören-Pürsün E, Banerji A, Bernstein JA, Balle Boysen H, Busse PJ, et al. Consensus on treatment goals in hereditary angioedema: A global Delphi initiative. J Allergy Clin Immunol. 2021;148(6):1526–32.
- Vincent D, Parsopoulou F, Martin L, Gaboriaud C, Demongeot J, Loules G, et al. Hereditary angioedema with normal C1 inhibitor associated with carboxypeptidase N deficiency. J Allergy Clin Immunol Glob. 2024;3(2):100223.
- Reshef A, Buttgereit T, Betschel SD, Caballero T, Farkas H, Grumach AS, et al. Definition, acronyms, nomenclature, and classification of angioedema (DANCE): AAAAI, ACAAI, ACARE, and APAAACI DANCE consensus. Journal of Allergy and Clinical Immunology.
- Zuraw BL. Hereditary angioedema with normal C1 inhibitor: Four types and counting. J Allergy Clin Immunol. 2018;141(3):884-5.
Santacroce R, D'Andrea G, Maffione AB, Margaglione M, d'Apolito M. The Genetics of Hereditary Angioedema: A Review. J Clin Med. 2021;10(9). - Cao Y, Liu S, Zhi Y. Recurrent and acute abdominal pain as the main clinical manifestation in patients with hereditary angioedema. Allergy Asthma Proc. 2021;42(2):131-5.
- Patel N, Suarez LD, Kapur S, Bielory L. Hereditary Angioedema and Gastrointestinal Complications: An Extensive Review of the Literature. Case Reports in Immunology. 2015;2015(1):925861.
DNK-AND-0001, 25/04/25